
Issue 3 — On the Blood Brain Barrier
A horizontal review
A clear understanding of the blood–brain barrier (BBB) is essential for elucidating the pathophysiology and therapeutic challenges of many neurological disorders. Over the coming weeks, I will be developing a series of focused reviews on this topic. This document serves as an introductory overview, outlining the fundamental architecture of the BBB, its pathological remodelling in disease, and emerging technological strategies for its modulation and bypass. Each theme introduced here will be expanded in subsequent instalments.
Molecular and biophysical barrier architecture of the blood brain barrier
The blood–brain barrier (BBB) is best understood as a multi-scale, emergent property of the neurovascular unit (NVU): a composite of brain endothelial cells (BECs) whose plasma membranes, intercellular junctional complexes, polarized transportome, perivascular basement membranes, glycocalyx, pericytes and astrocyte endfeet interact to produce extraordinarily low paracellular conductance, tightly regulated transcytosis, and asymmetric transporter expression.
Perhaps one of the most important conceptual advances in BBB biology over the past few decades, has been the recognition that it is not a static anatomical structure but rather is a dynamic interface, constantly being tuned in response to mechanical forces (shear, tangential etc), internal metabolic cues and microenvironmental signals [1, 2]. The molecular constituents and functions of the BBB may be summarised with the following points. These points provide a basic conceptual framework from which the molecular and biophysical architecture of the BBB can be systematically explored and expanded.
i. Junctional complexes constitute the paracellular gate of the BBB [3, 4]
ii. The brain endothelium suppresses bulk fluid-phase transcytosis and relies on tightly regulated receptor and carrier mediated transportation [5-8]
iii. Perivascular cells alongside astrocytes provide trophic signalling and mechanical stabilisation necessary for endothelial differentiation [8-11]
iv. There is overlap and reciprocity in all these functions such that they are integrated into a single responsive continuum that conserves neurovascular homeostasis [12, 13]
Junctional complexes and regulation of paracellular flux at the BBB
The paracellular barrier of the BBB arises from a tightly (no pun) integrated network of intercellular junctions. Principally, these are the tight junctions (TJs) and adherens junctions (AJs) that act in concert to restrict solute flux, preserve endothelial polarity, and coordinate mechanical and chemical signalling within the neurovascular unit.
VE-cadherin (aka cadherin5 / CDH5) is the canonical component of AJs, forming strand swapped adhesive dimers through their membrane-distal EC1 domains in which N terminal β strands of opposing molecules are reciprocally exchanged across adjacent endothelial cells (figure 1) [14]. This Ca²⁺-dependent homophilic interaction is stabilized by five extracellular cadherin repeats (EC1–EC5) that rigidify the intercellular interface. Intracellularly, CDH5’s cytoplasmic tail binds β-catenin, α-catenin, and p120-catenin, which tether the complex to cortical F-actin and dynamically regulate junctional tension and mechanotransduction [15, 16].


Claudin-5 (CLDN5) is the principal structural determinant of tight junctions and BBB integrity as a whole. Thus, genetic ablation of Cldn5 causes neonatal lethality with brain oedema, whereas overexpression increases TEER in vitro [4]. CLDN5 is a part of the tetraspan claudin family whose extracellular loops (ECL1 and ECL2) mediate cis- and trans-interactions that define TJ strand continuity and charge selectivity [17, 18]. The. claudin C-terminal PDZ-binding motifs recruit ZO-1, ZO-2, and ZO-3 thereby establishing the link to intracellular actin [19] (see figure 2).
The crosstalk between VE-cadherin and claudin-5 integrates AJs and TJ signalling within the brain endothelium[20]. In vitro, VE-cadherin clustering promotes upregulation of claudin-5 via modulation of FoxO1 transcriptional activity, acting through the β-catenin/FoxO1 axis. VE-cadherin engagement sequesters β-catenin at the membrane, thereby limiting its nuclear translocation and transcriptional repression of CLDN5[20]. Consequently, loss of VE-cadherin or disruption of its adhesive interface leads to β-catenin nuclear accumulation, FoxO1 activation, and downregulation of claudin-5, coupling adherens junction integrity to tight junction gene expression and barrier stability.
Endocytic control of transcellular transport at the BBB
Endocytosis within BECs is largely degradative in nature yet remains functionally significant for maintaining homeostatic control of transcellular transport. Clathrin-mediated endocytosis (CME) constitutes the principal mechanism for receptor internalisation at the BBB, whereas caveolae-dependent uptake which is dominant in peripheral endothelia is transcriptionally and functionally repressed[7]. The regulatory dichotomy between CME and caveolar pathways underpins much of the BBB’s selective transport competence. This section therefore focuses on these two paradigmatic mechanisms. For a more in depth discussion I refer the reader to forthcoming correspondences.
Ben-Zvi et al. identified the orphan solute carrier Mfsd2a as a critical determinant of BBB formation in mouse embryos, with its expression being approximately 80-fold higher in cortical endothelial cells compared to non-barrier endothelia. They also showed that genetic ablation of Mfsd2a resulted in pronounced BBB leakiness, evidenced by tracer extravasation from embryonic development through to adulthood, despite the preservation of tight junction ultrastructure [6]. Thus, given the preservation of paracellular integrity despite increased permeability, it follows that the Mfsd2a⁻/⁻ endothelium suffers from a defect in transcellular regulation rather than junctional formation. Hence, Mfsd2a functions not as a structural determinant of tight junctions, but as a negative regulator of vesicular transcytosis.
Key point : Mfsd2a is primarily responsible for caveolae-dependent uptake repression in developing BBB
Andreone et al. (2017) then demonstrated that Mfsd2a suppresses caveolae formation via its lipid transport function. They found that heterologous expression of Mfsd2a in non-brain endothelial cells was sufficient to reduce caveolar pit density and block uptake of albumin and transferrin. Conversely, loss of Mfsd2a increased tracer uptake and vesicle number even in otherwise quiescent brain endothelium. To dissect whether this reflected a structural or metabolic role, they generated a transport-deficient mutant (Mfsd2a D96A), which lacks sodium-dependent lysophosphatidylcholine (LPC) transport activity but remains stably expressed at the plasma membrane. Importantly, this mutant reproduced the knockout phenotype increasing caveolae formation and tracer permeability and thereby proving that Mfsd2a’s barrier function arises from its role in lipid transport rather than from any structural or protein scaffolding capacity [7].
Then they did a lipodomic analysis and observed wild-type brain microvessel enrichment in DHA-containing phospholipids, notably LPC-DHA derivatives, whereas Mfsd2a-deficient vessels showed reciprocal depletion of these species and accumulation of plasmenylethanolamines, which favour ordered lipid raft domains. DHA incorporation into phospholipids is known to reduce membrane order and disrupt caveolin-1 clustering [21]. Thus in this way they showed through correlation that by importing LPC-DHA into endothelial membranes, Mfsd2a alters bilayer composition to create an environment hostile to caveolae nucleation. If Mfsd2a activity defines this lipid signature, and DHA suppresses the formation of caveolin-rich microdomains, then Mfsd2a acts as a lipid-dependent governor of membrane architecture that passively represses caveolar biogenesis.
Furthermore, and perhaps more significantly, deletion of caveolin-1 in Mfsd2a⁻/⁻ mice completely rescued BBB permeability, normalizing tracer retention and vesicle density. Therefore, Mfsd2a operates upstream of Cav-1 in a hierarchical pathway that maintains low vesicular density in brain endothelium. While this suppression of transcytosis was restored, Mfsd2a⁻/⁻; Cav-1⁻/⁻ double mutants remained microcephalic, indicating that Mfsd2a’s lipid transport activity also serves an independent metabolic role in neuronal DHA supply [7].
Proper interaction between EC basement membranes, pericytes and astrocytes is required for the maintenance of the BBB's integrity
The cerebrovascular basement membrane (BM) is bi-layered (figure 3), comprising an endothelial (abluminal) layer and a parenchymal layer formed by astrocytic endfeet fusion. Molecularly, the BM is dominated by laminins, collagen IV, nidogens and heparan sulphate proteoglycans (e.g. perlecan and agrin). Importantly, the laminin isoforms from each BM layer are not interchangeable [22]. Thus, distinct α-chain compositions of endothelial (α4/α5) versus astrocyte (α2) laminins generate unique adhesive cues and receptor signalling that orchestrates junctional maturation, pericyte attachment, and barrier tightening. Altering this laminin complement remodels integrin and dystroglycan signalling, perturbs pericyte attachment and weakens endothelial tight junctions [23].
Pericytes are embedded within the endothelial BM and reciprocally shape its composition, thereby, by extension, affect BBB integrity [8, 24]. When cultured on extracellular matrix substrates, pericytes assemble laminin and collagen networks. They also secrete matrix metalloproteinases (MMPs) which degrade ECM components and modulate BM remodelling. In this way, pericytes dynamically affect the BM and consequently, the BBB. Beyond their role in matrix production, pericytes also present surface receptors which transduce matrix derived signals to modulate endothelial cell behaviour [25]. In fact, in Pdgfrb^-/- or Pdgfb^ret/ret mutants, where pericyte coverage is severely reduced, endothelial cells exhibit persistent vesicular activity and immature barrier properties, indicating that pericytes repress caveolar transport pathways while permitting junctional maturation [8, 9, 26]. This process is tightly linked to angiopoietin–Tie2, TGF-β, and Notch signaling, reflecting the central role of pericytic TGF-β–Ang2 cross-talk in maintaining endothelial quiescence and vascular stability [27].

Astrocytic endfeet deposit unique BM components (agrin, specific laminins), express receptors (α-dystroglycan, integrins) that stabilise endfoot adhesion, and modulate BM stiffness and composition through secretion of ECM enzymes and matricellular proteins [28]. Astrocyte-derived BM constituents are critical for establishing the parenchymal BM that enforces polarity of transporters and pumps on the abluminal endothelial membrane. Indeed, astrocyte loss or altered astrocytic signalling rewires BM composition and accelerates barrier breakdown in disease [29].
Pathophysiological modulation of the BBB in disease
Most of the topics discussed here will be the subject of future letters in this series so I will try to be brief.
The disruption of the BBB in neurodegenerative disease and stroke : Immune cell trafficking and the neuroimmune axis
BBB breakdown is both an early and progressive feature of neurodegenerative disease. In alzheimers for example, there is a loss of pericyte-endothelial crosstalk which leads to the upregulation of endothelial transcytosis as well as degradation of cldn-5 and occludins [30]. The resulting permeability allows plasma proteins, such as fibrinogen and immunoglobulins to extravasate into the perivascular and parenchymal space, triggering microglial activation and neuroinflammation. In fact, ApoE4 alleles which are the strongest genetic risk factor for late onset sporatic AD act in part through this pathway[31, 32]. ApoE4-expressing pericytes activate the cyclophilin A–NF-κB–MMP9 cascade, degrading basement membrane components and further weakening junctional complexes [33]. Similar processes are at play in Parkinson’s disease and multiple sclerosis although the initiating mechanisms may differ. In PD, α-synuclein aggregation disrupts endothelial tight junction organization, increasing permeability particularly in the nigrostriatal vasculature. In MS, BBB breakdown enables autoreactive lymphocyte infiltration via upregulation of endothelial adhesion molecules (VCAM-1, ICAM-1) and chemokines (CCL2, CXCL12), marking the transition from a relatively immune-privileged to a pro-inflammatory vascular phenotype [34]. These alterations are sustained by cytokines such as TNF-α and IFN-γ that remodel endothelial cytoskeletons and junctions via RhoA and PKC signalling pathways.
For good review of BBB dynamics in PD see [35]
Acute ischemic stroke is a bit different. BBB disruption occurs temporally. In the hyperacute phase, ischemia-induced hypoxia and energy failure cause cytoskeletal contraction and early tight junction disassembly. Reactive oxygen species (ROS) and matrix metalloproteinases (especially MMP-2 and MMP-9) degrade basement membrane components, resulting in vasogenic oedema and secondary neuronal injury [36]. The post-ischemic inflammatory phase recruits leukocytes that exacerbate permeability via release of TNF-α and IL-1β. The good news is that this transient window of permeability is therapeutically exploitable as it permits enhanced delivery of neuroprotective or thrombolytic agents into the penumbral tissue.
Loss of trophic coupling in the NVU in vascular dementia
Vascular dementia (VaD) is increasingly recognized as a disease of neurovascular uncoupling, where chronic cerebrovascular insufficiency and endothelial dysfunction lead to progressive neuronal loss [37, 38]. Chronic hypoperfusion, as seen in small vessel disease, triggers endothelial oxidative stress and reduces nitric oxide (NO) bioavailability through the upregulation of NADPH oxidases and eNOS uncoupling [39]. Reduced NO signaling impairs vasodilation and suppresses endothelial secretion of neurotrophic mediators e.g. brain-derived neurotrophic factor (BDNF) and platelet-derived growth factor-B (PDGF-B) [39, 40]. Consequently, pericyte survival and contractile function decline, weakening BBB integrity and reducing microvascular autoregulation [41]. There is also astrocytic dysfunction. Normally, astrocytes regulate local blood flow in response to neuronal activity through Ca²⁺-dependent release of vasoactive metabolites (prostaglandins, epoxyeicosatrienoic acids). In hypoperfused states, reactive astrocytes lose this precision and instead release endothelin-1 and cytokines, further impairing microvascular tone and promoting endothelial-to-mesenchymal transition (EndoMT)[42] [43]. Meanwhile the loss of astrocytic lactate shuttling and diminished AQP4 polarisation disrupts neuronal energy support, thus compounding cognitive decline [44].
At the ultrastructure level, perivascular fibrosis and thickening of the basement membrane reduce oxygen and nutrient exchange. Endothelial tight junctions are often retained but functionally leaky, reflecting altered claudin and ZO-1 phosphorylation states rather than overt degradation [45]. These cumulative changes drive a “silent” BBB leak, permitting low-grade plasma extravasation and chronic microglial activation, which together may accelerate white matter rarefaction and neuronal death [46].
Thus, in VaD, the fundamental lesion lies not in overt barrier collapse but in the gradual metabolic and trophic uncoupling of NVU constituents. Endothelial and glial signals lose reciprocity, neurons receive insufficient trophic support, and barrier homeostasis deteriorates secondary to energetic failure.
The blood-tumour barrier : Quasi-decimation of the BBB
The blood–tumour barrier (BTB) represents a pathological remodelling of the BBB in the context of primary and metastatic brain tumours. Unlike the systemic vasculature of most tumours, the BTB retains partial barrier characteristics, producing a heterogeneous permeability landscape that both impedes drug delivery and sustains tumour progression [47].
At the cellular level there is a downregulation of CLDN-5 and occludin expression, as well as a redistribution and phosphorylation of VE-cadherin. Thus cell-cell adhesion is compromised [48]. Pericyte coverage becomes irregular, with frequent detachment driven by VEGF-A and angiopoietin-2 signaling, leading to regions of high leak interspersed with relatively impermeable capillaries [49]. The resulting vessel architecture is tortuous, dilated, and poorly perfused, with variable hydrostatic pressures across the tumour core.
Astrocytic endfoots also become displaced from the vascular surface due to glial retraction and extracellular matrix remodelling. This loss of astrocytic contact disrupts paracrine signalling, particularly reduced Sonic hedgehog and angiopoietin-1 delivery, further destabilizing endothelial phenotype [47]. Simultaneously, tumour-derived cytokines such as IL-8 and TGF-β promote endothelial dedifferentiation and endothelium-to-mesenchymal transition, generating a quasi-barrier endothelium with impaired polarity and increased vesicular transport.
There is also Mfsd2a expression suppression in BTB endothelium, resulting in de-repression of caveolae-mediated transcytosis and enhanced albumin and drug leakage [50]. Interestingly, this permeability is spatially heterogeneous with perivascular regions adjacent to viable tumour cells often re-establishing partial tight junctions and reducing vesicular activity, thus creating microdomains of restricted diffusion. Consequently, therapeutic agents encounter inconsistent penetration, limiting efficacy of chemotherapy and biologics [47].
Examples of nanotechnologies being employed to bypass / modulate the BBB
Lactoferrin-coated COOH-PEG-PAsp-PV@Se micelles
A polymeric micelle is an equilibrium self-assembly of amphiphilic block copolymers into core–shell nanoparticles
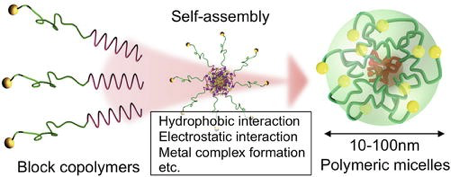
Amphiphilic block copolymers can self-assemble into micelles in selective solvents, forming soft, compressible spheres with a compact, insoluble core and an elastic corona [51]. When deposited as monolayers, these micelles tend to arrange in hexagonally ordered arrays resembling close-packed hard spheres. Micelles can also serve as nanocompartments for controlled loading of inorganic precursors. Following removal of the polymer matrix (e.g., by plasma treatment), the encapsulated inorganic salts can be converted into noble metal or metal oxide nanoparticles, preserving the spatial order of the original micellar template [52]. The final arrangement, clusters of nanoparticles organized in regular hexagonal arrays, depends critically on the precision of the micellar deposition process.
In COOH–PEG–PAsp–PV@Se, the hydrophobic PV@Se core sequesters poorly water-soluble drugs and introduces redox-responsive selenium functionalities, while the poly(aspartic acid) (PAsp) and poly(ethylene glycol) (PEG) blocks constitute the hydrophilic corona, conferring colloidal stability and surface chemistry amenable to bioconjugation. The carboxyl termini (–COOH) of PAsp or PEG act as coupling sites for ligands such as lactoferrin (Lf) via EDC/NHS activation, forming amide bonds to lysine residues without perturbing protein tertiary structure
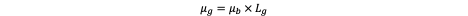
Under physiological conditions PAsp’s carboxylates are deprotonated (–COO⁻), ensuring electrostatic repulsion and hydration; in the mildly acidic endosomal milieu (pH 5–6) protonation diminishes repulsion, allowing micellar destabilization and accelerated payload release. Thus, PAsp also imparts a pH-responsive character.
Selenium plays a dual role within this system, functioning both as an intrinsic antioxidant as well as a redox-responsive trigger (see above figure). This apparent therapeutic synergy belies the elegance of this design, particularly for the treatment of oxidative stress-driven neurodegenerative disorders like Alzheimer’s disease. The selenium-containing domains are readily oxidizable, enabling controlled modulation of micellar polarity. Upon oxidation, these domains alter the hydrophilic–hydrophobic balance of the copolymer, leading to micellar destabilization and disassembly. This redox-driven structural transition provides a precise mechanism for triggered drug release within pathological microenvironments enriched in ROS. In addition to this, selenium also participates in catalytic redox cycles that can scavenge H₂O₂ and other ROS by reversible redox interconversions (Se²⁻ ↔ Se⁰ ↔ Se⁴⁺ in various organic contexts). As such, applying this mechanism to the treatment of neurodegenerative diseases like alzheimers where there is a oxidative microenvironment, allows for both targeted drug delivery and neutralisation of the oxidative environment.
Targeted drug delivery is further achieved by lactoferrin coating. This functionalisation adds a biologically precise vector for receptor mediated transcytosis. Lactoferrin is an iron-binding glycoprotein which binds to lactoferrin receptors (LfR) (part of the transferrin family) abundantly expressed on brain microvascular endothelial cells. Lf conjugation enables selective engagement of RMT pathways, promoting micellar endocytosis and transcytosis across the BBB. Its moderate binding affinity favors receptor recycling rather than lysosomal degradation, while PEG spacing maintains epitope accessibility.
Ouyang et al (2023) were able to integrate all these functionalities and showed that LfH-PIC@Se micelles are coherently capable of addressing alzheimer’s pathology at multiple levels (figure 6) [54]. In vitro, LfH–PIC@Se attenuated H₂O₂-induced mitochondrial depolarization in PC12 neuronal cells, confirming ROS scavenging and mitochondrial protection (figure II). Hemocompatibility assays showed hemolysis rates below 1%, underscoring excellent biocompatibility relative to conventional nanomaterials. In vivo fluorescence imaging in mice revealed markedly enhanced brain accumulation of CyP-loaded LfH–PIC@Se compared to free CyP, confirming effective BBB penetration mediated by LfR interaction. Thus, with their findings Ouyang et al were able to substantiate this framework as a promising strategy for redox-modulated, receptor targeted drug delivery in AD.
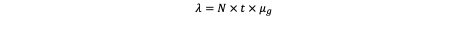
Retrograde axonal delivery with neural tracer conjugated nanoparticles
Rather than modulate/penetrate the BBB, one could alternatively, bypass it entirely by utilising neuronal axonal transport as a delivery highway. Payloads applied at distal nerve terminals can be conveyed retrogradely to neuronal cell bodies [55]. As a honorary mention, Mao’s group at UNSW have advanced this paradigm by conjugating wheat germ agglutinin–horseradish peroxidase (WGA-HRP), a classical neural tracer with strong retrograde transport capacity, onto gold nanoparticle (AuNP) cores, thereby imparting axonal targeting and transport functionality [56]. WGA binds to neuronal surface glyco-motifs (e.g. N-acetylglucosamine, sialic acid) and is internalized via clathrin-mediated endocytosis, then trafficked by dynein motors along microtubules. By chemically conjugating WGA-HRP to AuNP, the nanoparticle inherits the tracer’s intrinsic capacity for retrograde sorting and transport. The conjugation is covalent (via classical coupling chemistries), preserving ligand orientation and function [57].
Thus, in their work, Mao et al. cultured primary dorsal root ganglion (DRG) neurons in microfluidic compartmental devices separating axonal termini from somata and thus enabling controlled exposure and real-time imaging of transport dynamics. Without the tracer, ~55.8 % of AuNP-containing endosomes were stationary, 39.5 % underwent retrograde motion, and a minor fraction showed anterograde motion. In contrast, AuNP–WGA-HRP endosomes preferentially underwent retrograde movement (~83.7 %) with reduced stationary dwell times. The average retrograde velocity was ~1.4 ± 0.5 µm/s for AuNP–WGA-HRP (vs ~0.5 ± 0.2 µm/s for bare AuNP, and ~1.1 ± 0.5 µm/s for WGA-HRP itself). They also analysed the “run-and-pause” behaviour along the axon and found that AuNP–WGA-HRP endosomes sustained longer runs, shorter pauses, and higher instantaneous velocities than unconjugated AuNP. Dynein inhibition (via dynarrestin) abolished retrograde motion of both AuNP–WGA-HRP and free WGA-HRP, confirming that transport was mediated by dynein-driven microtubule machinery.
If surface functionalization with WGA–HRP alters the interfacial chemistry of AuNPs, then it must also redefine their intracellular trafficking. This conjugation indeed “activated” rapid, directional retrograde transport otherwise absent in bare nanoparticles, implying a shift in endosomal sorting toward retrograde vesicles and reduced stationary trapping. Hence, the authors proposed that WGA–HRP functionalization reprograms endosomal fate to favour axonal transport. Leveraging this, they conjugated therapeutic agents (e.g., pro–adenosine receptor antagonists) to AuNP–WGA–HRP constructs, achieving targeted delivery to spinal and brainstem neurons via peripheral administration and restoring diaphragm function at only ~0.1% of the systemic dose.
References
1. Santaguida, S., et al., Side by side comparison between dynamic versus static models of blood–brain barrier in vitro: A permeability study. Brain Research, 2006. 1109(1): p. 1-13.
2. Cucullo, L., et al., The role of shear stress in Blood-Brain Barrier endothelial physiology. BMC Neuroscience, 2011. 12(1): p. 40.
3. Furuse, M., et al., Occludin: a novel integral membrane protein localizing at tight junctions. The Journal of cell biology, 1993. 123(6): p. 1777-1788.
4. Nitta, T., et al., Size-selective loosening of the blood-brain barrier in claudin-5–deficient mice. The Journal of Cell Biology, 2003. 161(3): p. 653-660.
5. Nguyen, L.N., et al., Mfsd2a is a transporter for the essential omega-3 fatty acid docosahexaenoic acid. Nature, 2014. 509(7501): p. 503-506.
6. Ben-Zvi, A., et al., Mfsd2a is critical for the formation and function of the blood–brain barrier. Nature, 2014. 509(7501): p. 507-511.
7. Andreone, B.J., et al., Blood-Brain Barrier Permeability Is Regulated by Lipid Transport-Dependent Suppression of Caveolae-Mediated Transcytosis. Neuron, 2017. 94(3): p. 581-594.e5.
8. Armulik, A., et al., Pericytes regulate the blood–brain barrier. Nature, 2010. 468(7323): p. 557-561.
9. Daneman, R., et al., Pericytes are required for blood–brain barrier integrity during embryogenesis. Nature, 2010. 468(7323): p. 562-566.
10. Yao, Y., et al., Astrocytic laminin regulates pericyte differentiation and maintains blood brain barrier integrity. Nature Communications, 2014. 5(1).
11. Laterra, J., C. Guerin, and G.W. Goldstein, Astrocytes induce neural microvascular endothelial cells to form capillary‐like structures in vitro. Journal of Cellular Physiology, 1990. 144(2): p. 204-215.
12. Sweeney, M.D., S. Ayyadurai, and B.V. Zlokovic, Pericytes of the neurovascular unit: key functions and signaling pathways. Nature Neuroscience, 2016. 19(6): p. 771-783.
13. Mishra, A., et al., Astrocytes mediate neurovascular signaling to capillary pericytes but not to arterioles. Nature Neuroscience, 2016. 19(12): p. 1619-1627.
14. Brasch, J., et al., Structure and Binding Mechanism of Vascular Endothelial Cadherin: A Divergent Classical Cadherin. Journal of Molecular Biology, 2011. 408(1): p. 57-73.
15. Oas, R.G., et al., p120-catenin and β-catenin differentially regulate cadherin adhesive function. Molecular Biology of the Cell, 2013. 24(6): p. 704-714.
16. Barry, A.K., N. Wang, and D.E. Leckband, Local VE-cadherin mechanotransduction triggers long-ranged remodeling of endothelial monolayers. Journal of Cell Science, 2015. 128(7): p. 1341-1351.
17. Greene, C., et al., Microvascular stabilization via blood-brain barrier regulation prevents seizure activity. Nature Communications, 2022. 13(1).
18. Menard, C., et al., Social stress induces neurovascular pathology promoting depression. Nature Neuroscience, 2017. 20(12): p. 1752-1760.
19. Itoh, M., et al., Direct Binding of Three Tight Junction-Associated Maguks, Zo-1, Zo-2, and Zo-3, with the Cooh Termini of Claudins. The Journal of Cell Biology, 1999. 147(6): p. 1351-1363.
20. Gavard, J. and J.S. Gutkind, VE-cadherin and claudin-5: it takes two to tango. Nature Cell Biology, 2008. 10(8): p. 883-885.
21. Fuentes, N.R., et al., Membrane therapy using DHA suppresses epidermal growth factor receptor signaling by disrupting nanocluster formation. Journal of Lipid Research, 2021. 62: p. 100026.
22. Xu, L., A. Nirwane, and Y. Yao, Basement membrane and blood-brain barrier. Stroke and Vascular Neurology, 2019. 4(2).
23. Kang, M. and Y. Yao, Oligodendrocyte-derived laminin-γ1 regulates the blood-brain barrier and CNS myelination in mice. Cell Reports, 2024. 43(5): p. 114123.
24. Li, G., et al., The role of endothelial cell–pericyte interactions in vascularization and diseases. Journal of Advanced Research, 2025. 67: p. 269-288.
25. Fazio, A., et al., Signaling Role of Pericytes in Vascular Health and Tissue Homeostasis. International Journal of Molecular Sciences, 2024. 25(12): p. 6592.
26. Mäe, M.A., et al., Single-Cell Analysis of Blood-Brain Barrier Response to Pericyte Loss. Circulation Research, 2021. 128(4).
27. Dave, J.M., et al., Loss of TGFβ-Mediated Repression of Angiopoietin-2 in Pericytes Underlies Germinal Matrix Hemorrhage Pathogenesis. Stroke, 2024. 55(9): p. 2340-2352.
28. Milner, R., et al., The Rapid Decrease in Astrocyte-Associated Dystroglycan Expression by Focal Cerebral Ischemia is Protease-Dependent. Journal of Cerebral Blood Flow & Metabolism, 2008. 28(4): p. 812-823.
29. Cabezas, R., et al., Astrocytic modulation of blood brain barrier: perspectives on Parkinson’s disease. Frontiers in Cellular Neuroscience, 2014. 8.
30. Winkler, E.A., A.P. Sagare, and B.V. Zlokovic, The Pericyte: A Forgotten Cell Type with Important Implications for <scp>A</scp>lzheimer's Disease? Brain Pathology, 2014. 24(4): p. 371-386.
31. Troutwine, B.R., et al., Apolipoprotein E and Alzheimer's disease. Acta Pharmaceutica Sinica B, 2022. 12(2): p. 496-510.
32. Sanabria-Diaz, G., et al., Apolipoprotein E4 effects on topological brain network organization in mild cognitive impairment. Scientific Reports, 2021. 11(1).
33. Salloway, S., et al., Effect of APOE genotype on microvascular basement membrane in Alzheimer's disease. Journal of the Neurological Sciences, 2002. 203-204: p. 183-187.
34. Balasa, R., et al., Reviewing the Significance of Blood–Brain Barrier Disruption in Multiple Sclerosis Pathology and Treatment. International Journal of Molecular Sciences, 2021. 22(16): p. 8370.
35. Lau, K., R. Kotzur, and F. Richter, Blood–brain barrier alterations and their impact on Parkinson’s disease pathogenesis and therapy. Translational Neurodegeneration, 2024. 13(1).
36. Lakhan, S.E., et al., Matrix Metalloproteinases and Blood-Brain Barrier Disruption in Acute Ischemic Stroke. Frontiers in Neurology, 2013. 4.
37. Wang, F., et al., Dysfunction of Cerebrovascular Endothelial Cells: Prelude to Vascular Dementia. Frontiers in Aging Neuroscience, 2018. 10.
38. Lecordier, S., et al., Neurovascular Alterations in Vascular Dementia: Emphasis on Risk Factors. Frontiers in Aging Neuroscience, 2021. 13.
39. Moretti, R. and P. Caruso, Small Vessel Disease-Related Dementia: An Invalid Neurovascular Coupling? International Journal of Molecular Sciences, 2020. 21(3): p. 1095.
40. Janaszak-Jasiecka, A., et al., Endothelial Dysfunction Driven by Hypoxia—The Influence of Oxygen Deficiency on NO Bioavailability. Biomolecules, 2021. 11(7): p. 982.
41. Procter, T.V., A. Williams, and A. Montagne, Interplay between Brain Pericytes and Endothelial Cells in Dementia. The American Journal of Pathology, 2021. 191(11): p. 1917-1931.
42. Hostenbach, S., et al., The pathophysiological role of astrocytic endothelin-1. Progress in Neurobiology, 2016. 144: p. 88-102.
43. Shabir, O., J. Berwick, and S.E. Francis, Neurovascular dysfunction in vascular dementia, Alzheimer’s and atherosclerosis. BMC Neuroscience, 2018. 19(1).
44. Iliff, J.J., et al., A Paravascular Pathway Facilitates CSF Flow Through the Brain Parenchyma and the Clearance of Interstitial Solutes, Including Amyloid β. Science Translational Medicine, 2012. 4(147): p. 147ra111-147ra1.
45. Wardlaw, J.M., et al., Is Breakdown of the Blood-Brain Barrier Responsible for Lacunar Stroke, Leukoaraiosis, and Dementia? Stroke, 2003. 34(3): p. 806-812.
46. Zeng, Y., et al., Mechanisms Underlying Vascular Inflammaging: Current Insights and Potential Treatment Approaches. Aging and disease, 2025. 16(4): p. 1889.
47. Arvanitis, C.D., G.B. Ferraro, and R.K. Jain, The blood–brain barrier and blood–tumour barrier in brain tumours and metastases. Nature Reviews Cancer, 2020. 20(1): p. 26-41.
48. Selim, M.S., et al., Claudin 5 Across the Vascular Landscape: From Blood–Tissue Barrier Regulation to Disease Mechanisms. Cells, 2025. 14(17): p. 1346.
49. Cheng, L., et al., Glioblastoma Stem Cells Generate Vascular Pericytes to Support Vessel Function and Tumor Growth. Cell, 2013. 153(1): p. 139-152.
50. Xie, Y., et al., Wnt signaling regulates MFSD2A-dependent drug delivery through endothelial transcytosis in glioma. Neuro-Oncology, 2023. 25(6): p. 1073-1084.
51. Kuperkar, K., et al., Amphiphilic Block Copolymers: Their Structures, and Self-Assembly to Polymeric Micelles and Polymersomes as Drug Delivery Vehicles. Polymers, 2022. 14(21): p. 4702.
52. Hu, J., et al., Recent advances in non-ionic surfactant templated synthesis of porous metal oxide semiconductors for gas sensing applications. Progress in Materials Science, 2025. 150: p. 101409.
53. Kashyap, M., et al., Advances in Polymer Optimization for Enhanced Drug Delivery. 2023, Springer Nature Singapore. p. 27-51.
54. Ouyang, N., et al., Development of lactoferrin-coated multifunctional copolymer micelles to cross the blood-brain barrier. Drug Delivery and Translational Research, 2024. 14(3): p. 773-787.
55. Hassan, M.M., et al., Sustained A1 Adenosine Receptor Antagonist Drug Release from Nanoparticles Functionalized by a Neural Tracing Protein. ACS Chemical Neuroscience, 2021. 12(23): p. 4438-4448.
56. Wang, W., et al., Neural Tracing Protein‐Functionalized Nanoparticles Capable of Fast Retrograde Axonal Transport in Live Neurons. Small, 2024. 20(39).
57. Yong, J., et al., Interfacial Interactions between Neural Tracing Lectin–Gold Nanoparticle Conjugate and Cell Membrane Glycoproteins. Langmuir, 2025. 41(16): p. 10161-10176.